
本文对之前的分享有所删改,并增添了一些内容补充,本文较长,建议收藏相互印证,过柱子相关的大部分问题将在这里终结。以下为正文部分:
之前和师妹聊天,师妹传了一个合成群内的聊天记录,看到一个很有意思的问答,瞬间给笑惨了!
正好值研究生开学,有好几个同学私信菜籽,让出一期关于过柱子的方法技巧的分享。总结了许久,从历史讲下来,废话比较多,三易其稿,不能说多好,大概就全流程走一遍吧,供大家参考。
大家有补充的也可以后台私信分享,如果有必要,我们可以引用再做一期经验分享。
菜籽已经在化学行业摸爬滚打十四五年了,之前在药厂的时候一直觉得拿到一个化合物直接过柱子是一个很low的操作。
因为菜籽是工厂技术出身,所以拿到化合物的第一个想法就是能不能用结晶,打浆,减压蒸馏,酸碱洗等方法来纯化,如果不能的话还会考虑能不能转化上个保护基啥的,在其他步骤去纯化。
直到后来进到高校,做起来了毫克级别的反应,才发现是菜籽狭隘了,上述方法真的不适用,过柱子真的的很香。
而且目前在菜籽实验室来讲,过柱子的手段太多了,除了人力,还有过柱机,有循环制备型GPC,有高效液相制备,量少了偷个懒是完全可以的。

当然,像菜籽这样“特权的”人并不多,“ 普通人 ”想要用,是要求他们把传统柱子过好才可以预约的。

我们常说过柱子其实就是柱层析分离太阳成集团tyc45668cn,也叫柱色谱,1903年由俄国科学家首次发明使用,其把植物色素的溶液经过粉碎的碳酸钙粉末,发现从上到下在碳酸钙粉末上有不同的色带,通过对不同色带的分开提取,得到了较纯叶绿素,叶黄素等。
该方法经过经过百年的发展至今已经比较成熟,固定相也从单一的碳酸钙粉末发展到了氧化铝(又分为酸性、中性、碱性),硅胶,活性炭等,其中各大实验室最常用的便是硅胶,一般分为正相硅胶和反相硅胶!
固定相(硅胶)一般要经过纯化和活性处理,颗粒大小应当均匀统一,理论上其颗粒越小、同单位质量表面积越大,吸附能力就越高,分离效果也就越好。但是实际使用中固定相的颗粒太小时往往会存在两个情况:
二是颗粒小也会导致颗粒之间缝隙小,从而使溶剂的流速就太慢,耗时;以硅胶为例,目前已经商业化用量最大的硅胶尺寸为200-300目。
过柱手段这块,除了前面说的正向循环制备,高效液相制备,过柱机这些自动分析的设备,手动过柱方面主要分为常压过柱,减压过柱,加压过柱三种。其中:
减压过柱,因为洗脱剂流速过快,会损失大半部分塔板数,分离效果较差,适合分离TLC板子分的比较开的样品,尤其适合产物只有一个大极性杂质的样品;所有其大多数时间被用于化合物的预处理,过掉部分杂质后再仔细纯化。 相关内容见:
常压过柱是三者中分离效果最好的一种过柱方式,但是因为很多时候溶剂走的太慢,分离效果再好也会影响效率,所以这时候一般会给与压力,这就是所谓加压过柱;
加压过柱压力不宜过高,一般手动的用双链球,自动的就用个养鱼泵,两者都可以如图改装一下。

前面的内容算一些介绍,下面将以正相柱层析为例子,我们讲一下常规的上样、过柱流程:
选取柱子时,观察有无砂板,没有砂板的柱子记得底部填一些棉花,棉花不宜太厚也不宜太薄,能挡住硅胶不下来就好。这里面涉及到一个比较争议的问题,即甲醇过柱子会不会溶解硅胶?
配套的准备好最小极性的的洗脱剂,紫外灯(实验室个人推荐手提式,随时可以照一照),加压设备,以及试管架!
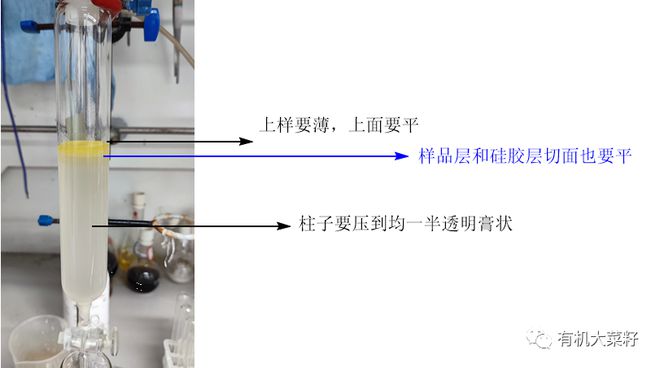
实验室最常见柱子直径和高度比值一般在1:2.5到1:15之间,上样时硅胶量是样品量的20~40倍左右。
选取柱子的标准要实际根据你样品的TLC爬板情况来,如果杂质很多挨得很近,那么尽可能选大一丢丢的柱子,使你的样品正好能铺一小薄层(建议厚度在5mm以下),如果杂质很远,这个厚度自然也可以提高,但也不建议你样品厚度超过2cm,除非这个产物就是简单冲一冲就行。
很多人会犯一个错误,就是想着我硅胶多加点,加的高高的,我样品上厚一点是不是也可以分离的很好?
理由是底下的样品和上面的样品不是在同一起跑线,这样只能的到一部分纯的,大部分是交叉的,过不纯不说,浪费时间浪费溶剂!
选取柱子后,加入硅胶时可以通过加料漏斗,同时一定要戴上口罩在通风橱里操作,原因是硅胶吸入人体后无法代谢,会将你的肺部打成筛子。硅胶粉是最毒的粉末之一,矽肺病约占全国尘肺病的50%!
②硅胶柱底部连接水泵,把硅胶抽实,然后从上面倒入极性最小的洗脱剂,到洗脱剂刚要漏下时,关闭截止阀,防止溶剂抽到水泵。
湿法上样,建议的做法是:用胶头吸管吸取样品,靠近硅胶最上面切面,缓慢均匀滴加样品,直到覆盖一层,然后停止加样,给点压力让油状物沉浸到硅胶层里;油状物沉浸到硅胶层里后重复这种操作多次,直至全部上完样!
有些新同学可能好奇为什么这样操作,而不是一次性把湿法上样的油状物一次性上完呢?
答案是这种操作可以减少样品层的有效高度。 即使还没有加洗脱剂,但当油状物浸入硅胶的那一刻,柱层析就已经开始,后面的样品相当于溶剂在推动前面的样品在硅胶里吸附、解吸附。
②干法上样就意味着样品层里会引入吸附剂,增加了样品层高度或柱子的尺寸,需要更多的时间,更多的溶剂。
如果不得已选择了,拌产物的硅胶用量一般以1~2倍样品量为宜。其上样直接把吸附好的粉慢慢倒进柱子就好,一些要求和湿法类似。
为了避免在加洗脱剂时撞击到样品层,使其不均匀,变形,影响分离效率,一般会在样品层上面铺一层无水硫酸钠或者石英砂。
菜籽个人喜欢铺无水硫酸钠,因为它还有干燥的功能,对于二氯甲烷这种易挥发吸热产生冷凝水的溶剂比较友好!

在上样之后,菜籽个人喜欢先用小极性溶剂(基本不会让产物下去的溶剂)冲柱2-3个柱体积,确保上下均一,然后选比例洗脱。
这是菜籽看到很多书中以及很多博主分享的过柱经验,实际过程中也大差不差,个人来讲更喜欢缩小一倍梯度洗脱。
比如如果TLC爬板展开溶剂是PE/EA=2:1,菜籽更可能从PE/EA=4:1开始梯度洗脱,洗脱到PE/EA=2:1结束,一般效果更好。
还有收集洗脱液的时候也是有技巧的,可以先选大一点的试管接收,TLC快到目标点了就换成小一点的试管,这样可以减少多接一点的可能性,不易包杂。

如果遇到杂质点和产物点挨得比较近的情况,特别是那些荧光又比较弱的,记得不要加压,应选常压过柱,每次接半管或更少就换管,也可以提高纯化几率。

很多新人有个习惯,过柱到了后期,TLC后面一两管没有产物,就默认柱子过完了,压干处理掉。
等到一计算收率才发现收率偏低,或者去打谱发现谱图不正确,抓错点了,后悔莫及。
所以菜籽建议您过完想要的点后柱子不要急忙处理,打核磁做收率都匹配之后再处理。
如果忙,或者急着用这根柱子纯化其他化合物,建议换大极性良性混合溶剂冲一下,把这部分洗脱液单独留下来等鉴定结果。
实际上,个人过柱的那些年,在看似出完产物的时候,都会换大极性溶剂冲一冲。遇到过有些东西一开始不了解形状,实际溶解性很差在柱子上断断续续出来,如用纯EA确定冲不出来了,但一旦换成二氯甲烷:甲醇体系,还会有!
展开剂是指你TLC爬板时的溶剂比例,洗脱剂是指你过柱时用的溶剂,理论上洗脱剂的极性不应大于展开剂的极性。
但某些特殊不好溶解的产物,过柱后期洗脱剂的极性可能会大些,常用洗脱剂极性以下面顺序递增:
对于没有砂板的柱子,填棉花有一个小技巧:可以直接拆一个衣架,用其将棉花投进入旋塞上面的出液口

过柱之前一定要留柱前样,特别是比较杂的体系;同时我也建议你过柱前TLC分析时点浓一点,有些杂质太稀的体系看不到!
菜籽看到很多硕士研究生这样操作,也看到过一些博士研究生这样操作,当时是懵逼状态,很不理解。
这得要多少的硅胶才能全部吸附住,本来固体只要1~2倍硅胶吸附,你这样操作最起码要十倍以上,也意味着相同柱子,柱效降低10倍以上,甚至远远不止!
如果你告诉我是因为产品固不固,液不液才这样选择,我会告诉你选方法重结晶试试!
举个最简单易懂的例子,擦完屁屁的卫生纸,再用你都会叠成两层,而且两层的情况下都有可能
一个是有些溶剂(如二氯甲烷、石油醚) 和硅胶混合的时候会明显放热,这也是为什么开始阶段有时候试管会发烫的原因,对温度敏感的化合物要小心;
另一个是甲醇会溶解不纯硅胶里的无机盐,不太建议用纯甲醇这种太大极性过柱子!
洗脱剂一般要根据展开剂比例选,但有时候PE/EA体系TLC不好时,可以用PE/DCM 体系试一试,说不定有更好的效果!
柱子下面的活塞记得一定不要涂硅脂之类的润滑剂,相似相溶原理,会被淋洗剂带到产品中从而导致杂峰;如果担心漏液,建议采用四氟节门的旋塞。

重要的事情说三遍。如果没有旋成粉状,成疙瘩颗粒就上样了,运气好可能是过不纯;运气不好可能就如同上图一样产物在柱子前头析出来,如果产品溶解性好也就罢了;如果溶解性特别不好,会导致整个柱子每个地方都有产品,即使用甲醇也一直冲不下来,到时候该怎么办?
硅胶对化合物的吸附性与化合物结构、官能团息息相关 ,一般含有极性较大的基团的化合物与硅胶吸附也越大,下面是根据个人经验总结的大概排序,可以参考一下:
碱和酸醇、胺、酰胺、硫醇、酚类酯、醛、酮硝基化合物>芳香族化合物卤代物、醚烯饱和烃
不要勉强也不要犹豫,最正确的做法是:直接换大极性溶剂直接冲下来,重新过柱,以节省时间。
如果过一些极性比较大的化合物如盐酸盐、碱性盐以及含有很多大极性基团的化合物,正相柱就不合适了,此时用到反向柱。

①有反向硅胶自然就有反相硅胶板,一般为铝板,两者价格都比较贵,所以要节省使用。
②反向柱体系可以类比HPLC,一般用甲醇-水体系,或乙腈-水体系过柱。过柱极性和正相柱相反,此体系中,甲醇大于水。
③反相柱不适用于全水体系,过完后反相硅胶要用甲醇冲洗干净,不要压干,用甲醇浸没保存
④反相柱也可以干法过柱,但拌样的的硅胶损坏比较厉害,内部空隙太大有缺陷,建议不要再回收。
两个作用,一个是抑制拖尾;一个是防止分解,特别是碱性化合物如亚胺,由于硅胶是酸性的,一定要小心。
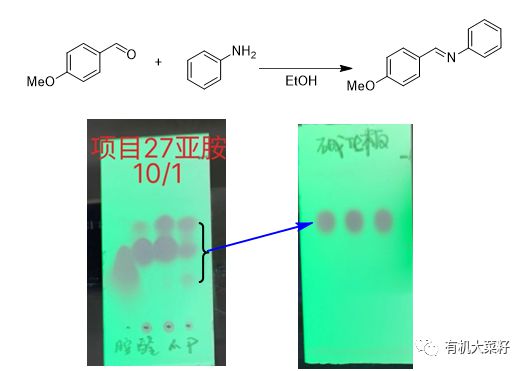
所以如果样品与硅胶的吸附比较强的话,就不容易流出,可能导致的后果就是,两个产物点,极性大的却更容易出来。
这种情况在有机合成中是经常遇到的。小编也遇到过多次,此时用氧化铝做固定相可以完美解决!
有些固体过柱后在溶液里TLC时点板是纯的,但是旋干点浓一点就会显现出杂质,相比较再过柱一遍,我更建议你打个浆,溶剂比例选择比洗脱剂极性小一点。
低级的酰氯肯定不适合,一定会分解;但是高级的固体酰氯还是比较稳定的,可以过柱子,但是我还是建议不要轻易接触水,毕竟水滴石穿!
一般不建议过夜,一方面硅胶是酸性的,可能会导致产物变坏,另一方面,溶剂在硅胶里面是非静止的的,在截止阀关闭后,无序运动会增大,可能导致产物的平面变得凌乱。
柱子干了这事比较可怕,柱子干了意味着硅胶里混进了很多气泡用来填充溶剂缺失的体积,直接导致塔板数降低,分离度降低!
听说给本文点赞,在看,喜欢的同仁们都要发 nature,science 了!!!




